
More Information
Submitted: 06 July 2019 | Approved: 26 August 2019 | Published: 28 August 2019
How to cite this article: Salem Y, Omar Y, Dorsaf S, Sonia M, Samir B, et al. The Pierre Marie-Sainton syndrome: Report of a family. J Oral Health Craniofac Sci. 2019; 4: 012-014.
DOI: 10.29328/journal.johcs.1001028
Copyright License: © 2019 Salem Y, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

The Pierre Marie-Sainton syndrome: Report of a family
Salem Y*, Omar Y, Dorsaf S, Sonia M, Samir B and Olfa B
Department of Pediatrics C, Children Hospital of Tunis, Faculty of Medicine of Tunis, Tunis El Manar University, Tunisia
*Address for Correspondence: Salem Yahyaoui, Department of Pediatrics C, Children Hospital of Tunis, Bab Saadoun, Tunisia, Tel: + 21671887713; Email: [email protected]
Pierre Maria and Sainton syndrome or cleido-cranial dysplasia (CCD) is a rare syndrome presenting an autosomal pattern of inheritance, characterized by characterized by a triad: clavicular aplasia, delayed ossification of the fontanelles and sutures of the vault of the skull. To these may be added multiple dental inclusions.
The Pierre Marie-Sainton syndrome is a rare dominant autosomal inherited skeletal disorder resulting from haploinsufficiency of the Runt-related transcription factor 2 (RUNX2) gene, a regulator for bone and cartilage development and maintenance [1,2]. It mainly affects bones that undergo intramembraneous ossification such as the calvarian and the clavicular bones with a wide range of expressivity [3-5]. It is also known as cleidocranial dysplasia (CCD), mutational dysostosis and cleidocranial dysostosis. It was first described by Pierre Marie and Paul Sainton in 1898 [6]. It is rare and only a few cases have been reported in the literature.
We aimed to contribute to the study of this exceptional affection by reporting three familial cases.
Case 1
Patient 1 was a male infant, born at 37 gestational weeks from non-consanguineous parents. He had no health problems until the age of 6 months when he was referred to our department for dyspnea. On physical examination, he had a normal weight for his age, his height was 62 cm. He had an obvious dysmorphism associating: an asymmetrical face, a bossing of the forehead, a depressed nasal bridge and a hypertelorism. The anterior and posterior fontanels were large with a disjunction of sutures. Both clavicles were hypoplastic. He had severe respiratory distress with inspiratory dypnea and dysphonia, Due to the serious clinical situation, the infant was referred to the intensive care unit with the diagnosis of asphyxiant laranygitis. After 5 days of mechanical ventilation, the evolution was favorable. The blood karyotype, performed for the etiological investigation of the dysmorphism, was normal. The transfontanellar ultrasound was normal. The skull X-ray showed open cranial sutures, delayed closure of fontanelles, multiple Wormian bones and poorly formed paranasal sinuses and zygomatic complex (Figure 1). Clavicular hypoplasia was confirmed by chest X-ray. Laryngoscopy showed laryngomalacia explaining the severity of respiratory distress. The diagnosis of Pierre Marie-Sainton syndrome was by the analysis of CBFA1 gene which identified a deletion mutation (220 del173).
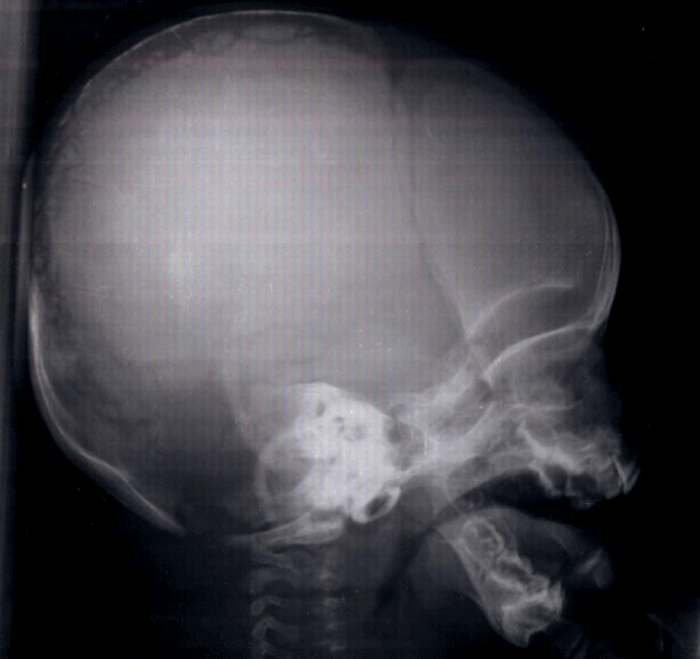
Figure 1: Skull radiography showing open skull sutures, large fontanels, multiple wormian bones, undeveloped paranasal sinuses.
Case 2
This it is the innate sister of the patient 1. She was three years old and she was doing well. She was examined as part of the family investigation. As her brother, she had large anterior and posterior fontanels and depressed nasal bridge. Chest X-ray showed bell shaped rib-cage, slender ribs and vertebral abnormalities (Figure 2). The diagnosis of cleidocranial dysplasia was confirmed by the identification of the same mutation as her brother.
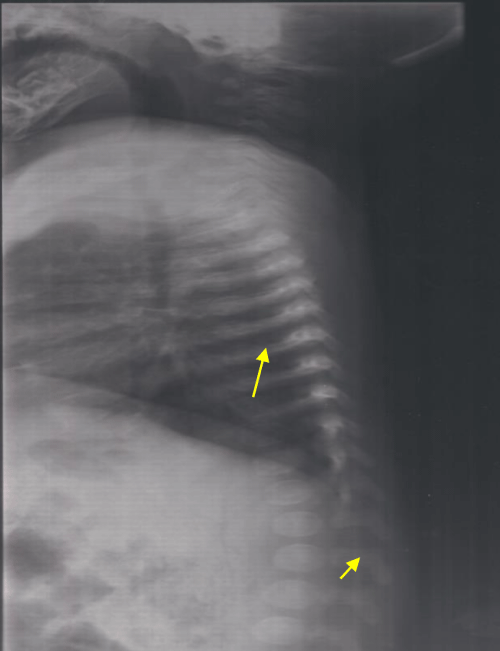
Figure 2: Lateral chest radiograph showing bell shaped rib-cage, slender ribs and vertebral abnormalities.
Case 3
The third case is the father of patients 1 and 2. He had no functional complaints. His physical examination showed a short stature with a height of 153 cm. He had facial dysmorphia with fronto-parietal bossing and depressed nasal bridge, prominent orbital ridges. He had also bilateral clavicular hypoplasia (Figure 3). Skull X-ray showed retention of many teeth and facial hypoplasia.
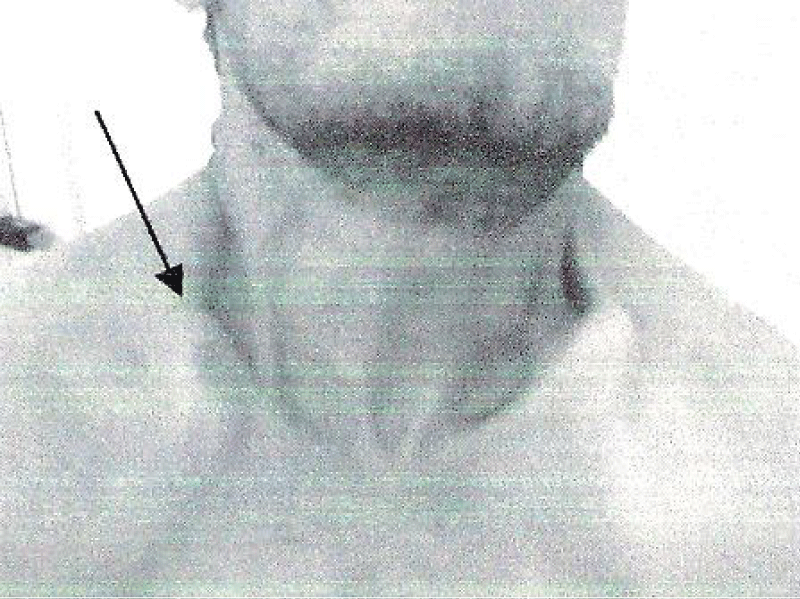
Figure 3: Bilateral clavicular hypoplasia.
Pierre Maria and Sainton syndrome is dominantly inherited skeletal dysplasia caused by mutations in the osteoblast-specific transcription factor CBFA1 [1]. It was first described by Marie and Sainton in 1898 [2]. The clinical spectrum is variable and ranges from mild CCD with isolated dental abnormalities which may be unnoticed to classic and severe cases [3-5]. The classic CCD phenotype is defined as the presence of hypoplastic clavicles and delayed closure of the anterior fontanelle in addition to craniofacial features. A genotype-phenotype correlation has been proven [1]. Moreover, the penetrance is variable and the clinical presentation is variable even in the same family as in the observations reported in this paper. The anterior fontanelle may remain open throughout adulthood [6-8]. Its patency associated with a wide-open metopic suture can produce a depression in the midline of the upper forehead, with a typical bulging aspect. Depressed nasal bridge and hypoplastic sinuses could disturb nasal breathing. Moreover, aplastic clavicles, deformed thorax and missing ribs may lead to respiratory insufficiency, especially in early infancy [9]. Therefore, CDD may cause more severe life-threatening disorder. Laryngeal involvement is a peculiarity of our first observation, which is reported for the first time. Bone X-ray is the easiest way to confirm the diagnosis [10]. The radiological signs are multiple. Dental abnormalities include unerupted teeth, narrow ascending ramus, cyst formation with supernumerary teeth and increased density of the alveolar crestal [11]. Skull X-rays can reveal brachycephaly, disjoint sutures, large fontanels and numerous wormian bones [10,12,13]. Other radiological abnormalities are possible such as vertebral defects, supernumerary ribs, scoliosis and pelvic bone abnormalities as well as hand and feet bones dysplasia [13].
Differential diagnosis of CDD includes hypohidrotic ectodermal dysplasia which includes hypohidrosis, abnormal dentition, onychodysplasia, and hypotrichosis, Apert syndrome characterized characterised primarily by craniosynostosis, mid-face hypoplasia, and syndactyly of the hands and feet, pyctodysostosis including a short stature, osteosclerosis and bone fragility [5,10,14].
Management is based on ventilatory support in case of respiratory complications, the correction of dental anomalies and reconstructive surgery [5,15].
Pierre Maria and Sainton syndrome is a rare entity. The clinical spectrum is wide. Although early diagnosis represents the main issue in the management of this syndrome, the clinical findings are often missed. Therefore, every practitioner should be aware of the major clinical characteristics to avoid diagnosis delay and to allow precocious. In confirmed cases, genetic counselling should be advised.
Consent
A written consent has been obtained from the patient for publication of this case report and accompanying images and is available for review on request.
Author contribution
Salem Yahyaoui and Dorsaf Saadouli wrote the paper
All authors provided care and follow-up for the patients
Olfa Bouyahya supervised the work.
Guarantor
Salem Yahyaoui has full responsibility for the work.
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
The authors described a familial hereditary case affecting father daughter and son.
- Lu Y, Li Y, Cavender AC, Wang S, Mansukhani A, et al. Molecular studies on the roles of Runx2 and Twist1 in regulating FGF signaling. Dev Dyn. 2012; 241: 1708-1715. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22972545
- Marie P, Sainton P. Sur la dysostose cleido-crânienne héréditaire. Rev neurol. 1898; 6: 835-838.
- Kallialia E, Taskinen PJ. Cleidocranial dysostosis: report of six typical cases and one atypical case. Oral Surg Oral Med Oral Pathol. 1962; 14: 808. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/14453327
- De Nguyen T, Turcotte JY. Cleidocranial dysplasia: review of literature and presentation of a case. J Can Dent Assoc. 1994; 60:1073-1078. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/7842373
- Golan I, Baumert U, Hrala BP, Müβig D. Dentomaxillofacial variability of cleidocranial dysplasia: Clinicoradiological presentation and systemic review. Dentomaxillofac Radiol. 2003; 32:347-54. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15070835
- Garg RK, Argawal P. Clinical spectrum of cleidocranial dysplasia: a case report. Cases J. 2008; 1: 377-381. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2614945/
- Nehta DN, Vachhani RV, Patel MB. Cleidocranial dysplasia: a report of two cases. J Indian Soc Pedod Prev Pent 2011 ; 29 : 251-254. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4869578/
- Thamzih Chelvan H, Malathi N, Vignesh Kailasam, Ponnidurai A. Cleidocranial dysplasia: a family report. Journal of Indian society of pedodontics and preventive dentistry. 2009; 4: 249-252. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19915277
- Brueton LA, Reeve A, Ellis R, Husband P, Thompson EM, et al. Apparent cleidocranial dysplasia associated with abnormalities of 8q22 in three indiciduals. Am J Med Genet. 1992; 43: 612-618. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/1605259
- Prakash R, Mohan S, Suma GN, Vashishth S, Goel S. Cleidocranial dysplasia: clinic-radiological illustration of a rare case. J oral science 2010; 1: 161-166. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4129351/
- Cooper SC, Flaitz CM, Johnston DA, Lee B, Hecht JT. A natural history of cleidocranial dysplasia. Am J Med Genet. 2001; 104: 1-6. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11746020
- Mc Namara CM, O’Riordan BC, Blake M, Sandy JR. Cleidocranial dysplasia: radiological appearances on dental panoramic radiography. Dentomaxillofac Radiol. 1999; 28: 89-97. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/10522197
- Tanaka JL, Ono Filho MH, Castilho JC, Moraes LC. Cleidocranial dysplasia: importance of radiographic imagines in diagnosis of this condition. J Oral Sci. 2006; 48: 161-166. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17023750
- Gonzalez Lopez BS, Ortiz Solalinde, Kubodera Itro T, Lara Carillo E, Ortiz Solalinde E. Cleidocranial dysplasia: report of a family. J Oral Sci. 2004; 46: 259-266.
- Roberts T, Stephen L, Beighton P. Cleidocranial dysplasia: a review of the dental, historical, and practical implications with an overview of the South African experience. Oral Med. 2013; 115: 45-55. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23102800